Sinais e sintomas
da PAF-TTR
Na PAF-TTR , as manifestações clínicas são multissistêmicas e debilitantes, com importante impacto psicológico e familiar.1,9
A doença pode ser influenciada pelo genótipo de TTR, pela localização geográfica ou por outros fatores genéticos e ambientais. Assim, a sintomatologia, a idade de início dos sintomas e a taxa de progressão variam entre os pacientes.
Os sintomas, que são inespecíficos e se assemelham a muitas condições, podem incluir neuropatia autonômica, disfunção gastrointestinal, manifestações oculares, distúrbios cardíacos, comprometimento da função renal ou síndrome do túnel do carpo.1,9
A neuropatia periférica sensitivo-motora é uma característica marcante da PAF-TTR ou Amiloidose Hereditária por Transtirretina com polineuropatia.
No início precoce (<50 anos de idade), pequenas fibras nervosas mielinizadas e não mielinizadas, que se associam à sensação de dor e temperatura são danificadas, o que pode se manifestar como parestesia, disestesia, alodinia, hiperalgesia ou dor espontânea nos pés. A degeneração axonal progride implacavelmente com padrão de distal a proximal.
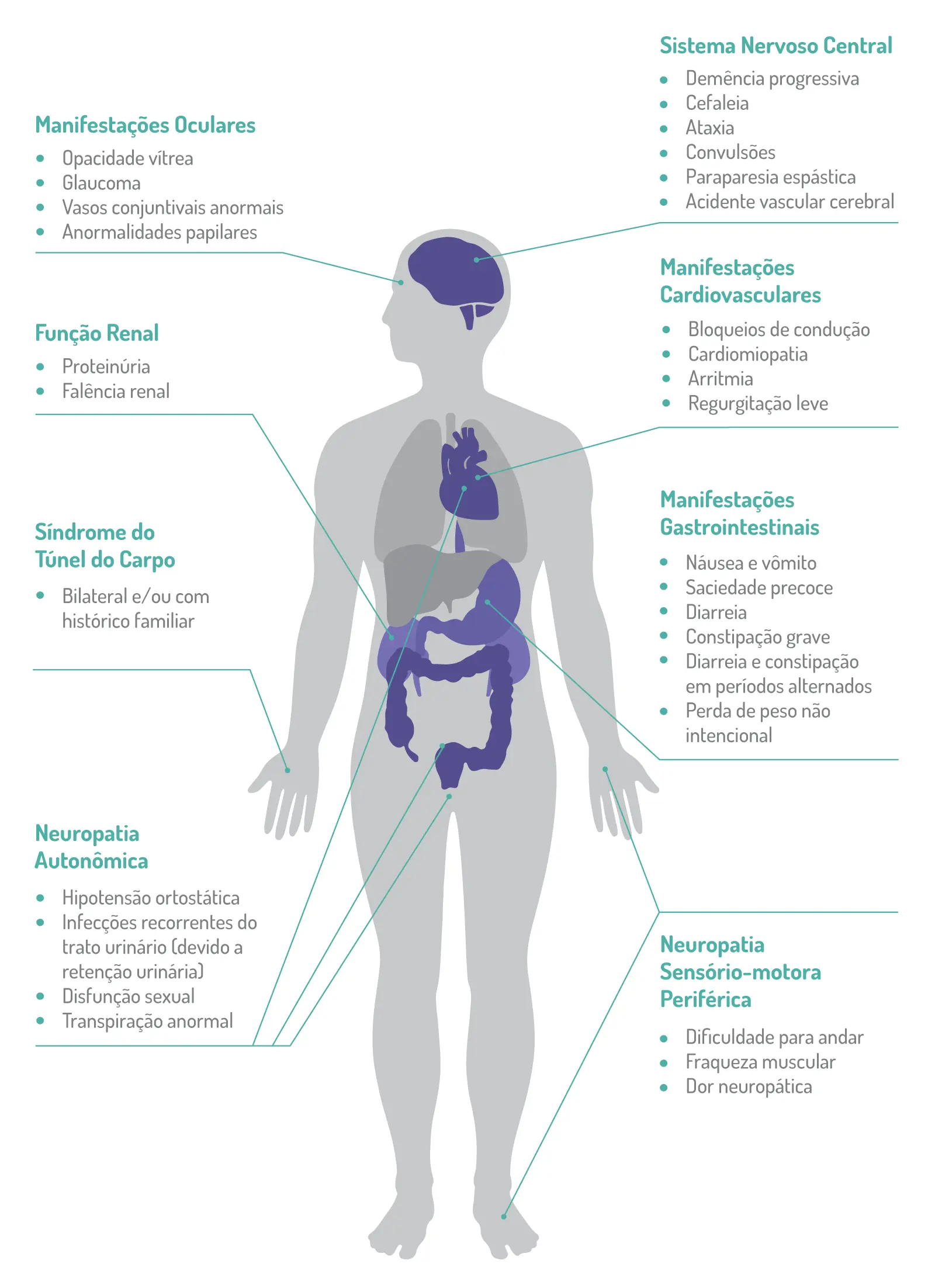
Após 2 anos, os pacientes apresentam sintomas motores, e depois de 4 a 5 anos aparecem os primeiros sintomas de nervos superiores, como síndrome do túnel do carpo bilateral nas mãos, que é bastante frequente. As deficiências motoras também progridem, aumentando a fraqueza e a dificuldade de andar. Os casos não tratados progridem para comprometimentos motores, autonômicos e sensoriais mais graves e podem levar à morte em aproximadamente 10 anos.5
Os casos de início tardio se caracterizam pela relativa preservação das fibras nervosas não mielinizadas e pela presença de brotamento axonal. Essas características são responsáveis por sensação (superficial e profunda) prejudicada, dor neuropática grave, comprometimento motor distal precoce e sintomas autonômicos relativamente leves.10
Estágios da PAF-TTR
Estágio 1
- Neuropatia sensorial e motora limitada aos membros inferiores;
- Dor e sensação térmica gravemente prejudicada, com leve toque e propriocepção relativamente poupada (dissociação sensorial);
- Comprometimento motor leve;
- Dificuldade de deambulação sem necessidade de bengala.
Estágio 2
- Necessidade do uso de bengala;
- Neuropatia progredindo para membros superiores e tronco;
- Comprometimento motor moderado.
Estágio 3
- Fase avançada;
- Paciente acamado ou em cadeira de rodas;
- Neuropatia sensorial, motora e autonômica grave em todos os membros.
Idade e gênero
As manifestações clínicas normalmente se desenvolvem entre 30 e 50 anos de idade e afetam igualmente homens e mulheres.
No entanto, a PAF-TTR com cardiomiopatia geralmente tem início tardio e é mais observada em pacientes com 60 anos de idade ou mais, principalmente em homens.
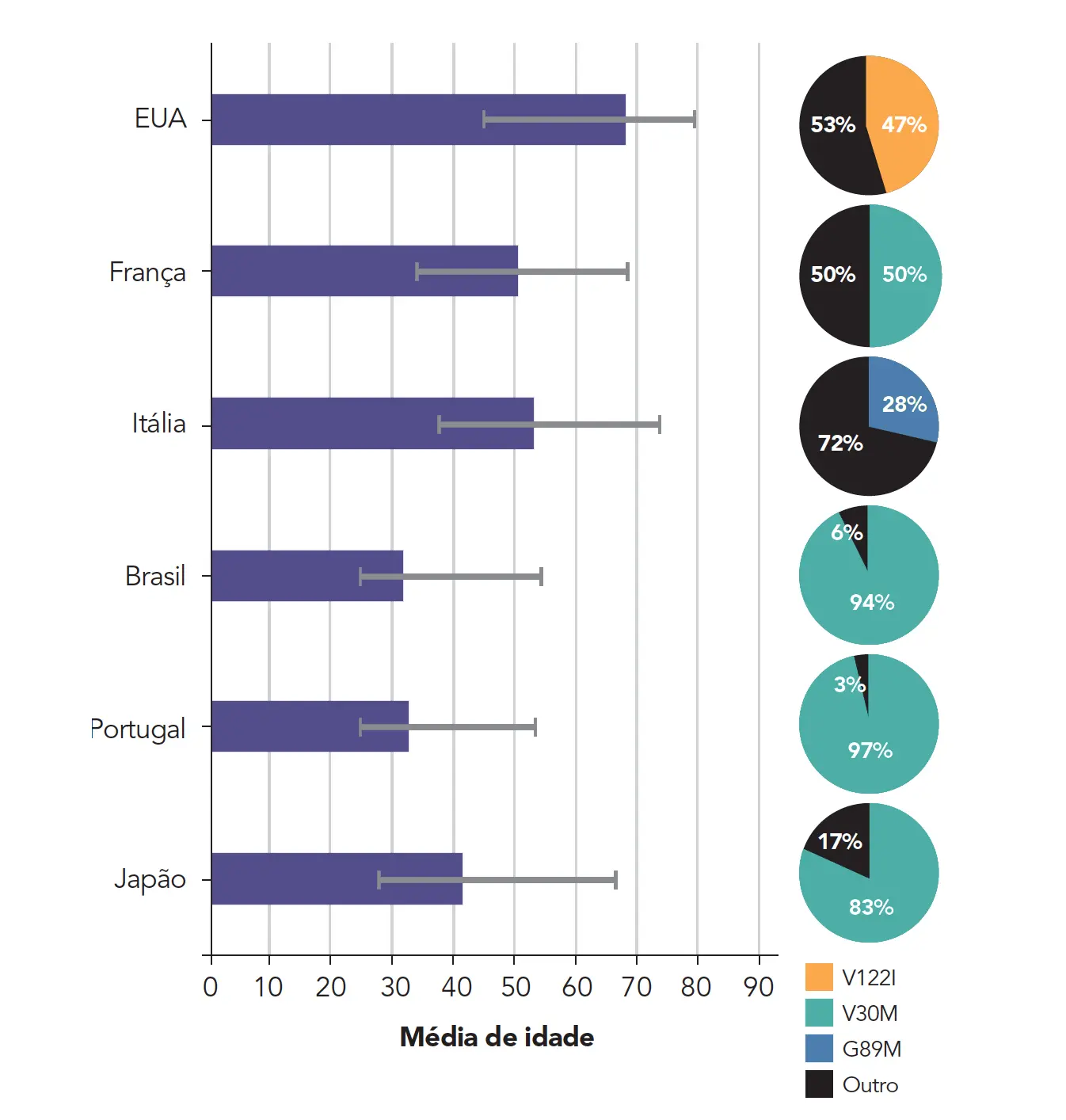
A idade média de início dos sintomas varia de acordo com o país e as mutações mais prevalentes:1
Estimativas recentes mostram prevalência mundial de 50 mil indivíduos, com diversas apresentações fenotípicas.1
Globalmente, a mutação mais comum da PAF-TTR é a V30M, seguida pela V122I.1
As mutações em V30M são predominantemente encontradas em Portugal, Espanha, França, Japão, Suécia e em países com descendentes dessas regiões, como o Brasil. As mutações em V122I são frequentemente observadas em afro-americanos.1
Prevalência da PAF-TTR
Um estudo sobre pacientes com neuropatia periférica de causa desconhecida, realizado no Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (HCFMRP-USP), identificou 129 mutações em 448 pacientes.
Na coorte de 128 pacientes de origem brasileira, observou-se que, embora a mutação V30M representasse 90,6% dos casos (semelhante ao panorama verificado nas áreas endêmicas como Portugal, Suécia e Japão), outras mutações também foram encontradas, devido à origem multivariada de nossa população, o que confirma o cenário de heterogeneidade genética no país.8
Referências Bibliográficas Clique para ver
1. Gertz MA. Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am J Manag Care. 2017 Jun;23(7 Suppl):S107-S112. Review. PubMed PMID: 28978215. /
2. Amyloidosis Support Groups. Conscientização sobre Amiloidose. Out. 2013. Disponível em: <http://amyloidosissupport.org/AmyloidAware_Portuguese.pdf> Acesso em 17 nov. 2018. /
3. COELHO, Teresa; MAURER, Mathew S; SUHR, Ole B. THAOS: The Transthyretin Amyloidosis Outcomes Survey: Initial Report On Clinical Manifestations In Patients With Hereditary And Wild-Type Transthyretin Amyloidosis. 2013. Disponível em: <https://www.ncbi.nlm.nih.gov/pubmed/23193944>. Acesso em: 21 ago. 2019 /
4. SCHMIDT, Hartmut H. et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle & Nerve, [s.l.], v. 57, n. 5, p.829-837, 1 fev. 2018. Wiley. http://dx.doi.org/10.1002/mus.26034. /
5. PINTO, Marcus Vinicius et al. Brazilian Consensus For Diagnosis, Management And Treatment Of Transthyretin Familial Amyloid Polyneuropathy. Arquivos de Neuro-psiquiatria, [s.l.], v. 76, n. 9, p.609-621, set. 2018. FapUNIFESP (SciELO). http://dx.doi.org/10.1590/0004-282×20180094. Disponível em: <https://www.ncbi.nlm.nih.gov/pubmed/30365625>. Acesso em: 21 ago. 2019. /
6. What does it mean if a disorder seems to run in my family? 2019. Disponível em: <https://ghr.nlm.nih.gov/primer/inheritance/runsinfamily>. Acesso em: 21 ago. 2019. /
7. RAPEZZI, Claudio et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. European Heart Journal, Oxford, v. 7, n. 34, p.520-528, 28 jun. 2012. Semanal. Disponível em: <https://academic.oup.com/eurheartj/article/34/7/520/598769>. Acesso em: 21 ago. 2019. /
8. LAVIGNE-MOREIRA, Carolina et al. The genetic heterogeneity of hereditary transthyretin amyloidosis in a sample of the Brazilian population. Journal Of The Peripheral Nervous System, [s.l.], v. 23, n. 2, p.134-137, 10 abr. 2018. Wiley. http://dx.doi.org/10.1111/jns.12259. Disponível em: <https://www.ncbi.nlm.nih.gov/pubmed/29520877>. Acesso em: 21 ago. 2019. /
9. LOPES, Alice et al. Life Paths Of Patients With Transthyretin-related Familial Amyloid Polyneuropathy: A Descriptive Study. Journal Of Community Genetics, [s.l.], v. 9, n. 1, p.93-99, 19 out. 2017. Springer Nature. http://dx.doi.org/10.1007/s12687-017-0338-0. Disponível em: <https://www.ncbi.nlm.nih.gov/pubmed/29052096>. Acesso em: 21 ago. 2019. /
10. CONCEIÇÃO, Isabel et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. Journal Of The Peripheral Nervous System, [s.l.], v. 21, n. 1, p.5-9, mar. 2016. Wiley. http://dx.doi.org/10.1111/jns.12153. Disponível em: <https://www.ncbi.nlm.nih.gov/pubmed/26663427>. Acesso em: 21 ago. 2019. /
11. Abstracts from the First European Meeting for ATTR /
12. Associação Brasileira de Paramiloidose. Disponível em: http://www.abpar.org.br. Acesso em: 20 ago. 2019 /
13. Sekijima Y. Hereditary Transthyretin Amyloidosis. 2001 Nov 5 [Updated 2018 Dec 20]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1194. Acesso em: 19/03/2020.
14. LANE, Thirusha et al. Quality of life in ATTR amyloidosis. Orphanet Journal of Rare Diseases. 2015; 10(Suppl 1):O26.
15. Amyloidosis Foundation. Understanding the patient voice in hereditary transthyretin-mediated amyloidosis (ATTR amyloidosis). Disponível em: http://amyloidosissupport.org/support_groups/fam_isabell_attr.pdf. Acesso em: 30 abr. 2020.
16. Stewart M, et al. Characterizing Disease Burden in an Ultra-Rare Disease in the United States: Transthyretin (TTR) Amyloidosis Patients & Caregivers. Value Health. 2013;16:A386 .